
Introduction
Next-Generation Sequencing of 16S ribosomal RNA (16S NGS) is a widely adopted method for profiling microbial communities across various sample types, including food, clinical, and environmental.1, 2, 3 By targeting the hypervariable regions such as V3-V4 or V4 within the highly conserved 16S rRNA gene, which is universally present in bacteria and archaea, this technique enables differentiation between taxa at the genus or species level.3 The success of next generation sequencing often hinges on the choice of the library prep protocol. Among the most recognized protocols are the Earth Microbiome Project (EMP) protocol,4 which targets the V4 region, and Illumina’s 16S NGS protocol,5 which targets the V3-V4 region. Here, we introduce Zymo Research’s line of Quick-16S™ Plus NGS Library Prep Kits, which provide comparable or superior results while offering a considerably streamlined workflow. Hands-on time has been reduced by as much as 8-fold, allowing researchers to prepare libraries quickly and easily, without compromising data quality.
Results and Discussion
To evaluate the performance of Zymo Research’s Quick-16S™ Plus NGS Library Prep Kit (V4) versus the EMP protocol (V4),4 we applied both methods to sequence the ZymoBIOMICS® Microbial Community DNA Standard as well as a variety of biological and environmental samples. The Quick-16S™ Plus NGS Library Prep Kit (V4) greatly simplifies the workflow, requiring only three simple steps compared to the four steps required by the EMP protocol (Figure 1). Notably, the EMP protocol requires additional DNA quantification and visualization steps, as well as pooling by equal mass for normalization purposes, all of which are labor-intensive and time-consuming, while the Quick-16S™ Plus kit completely foregoes these steps (Figure 1).
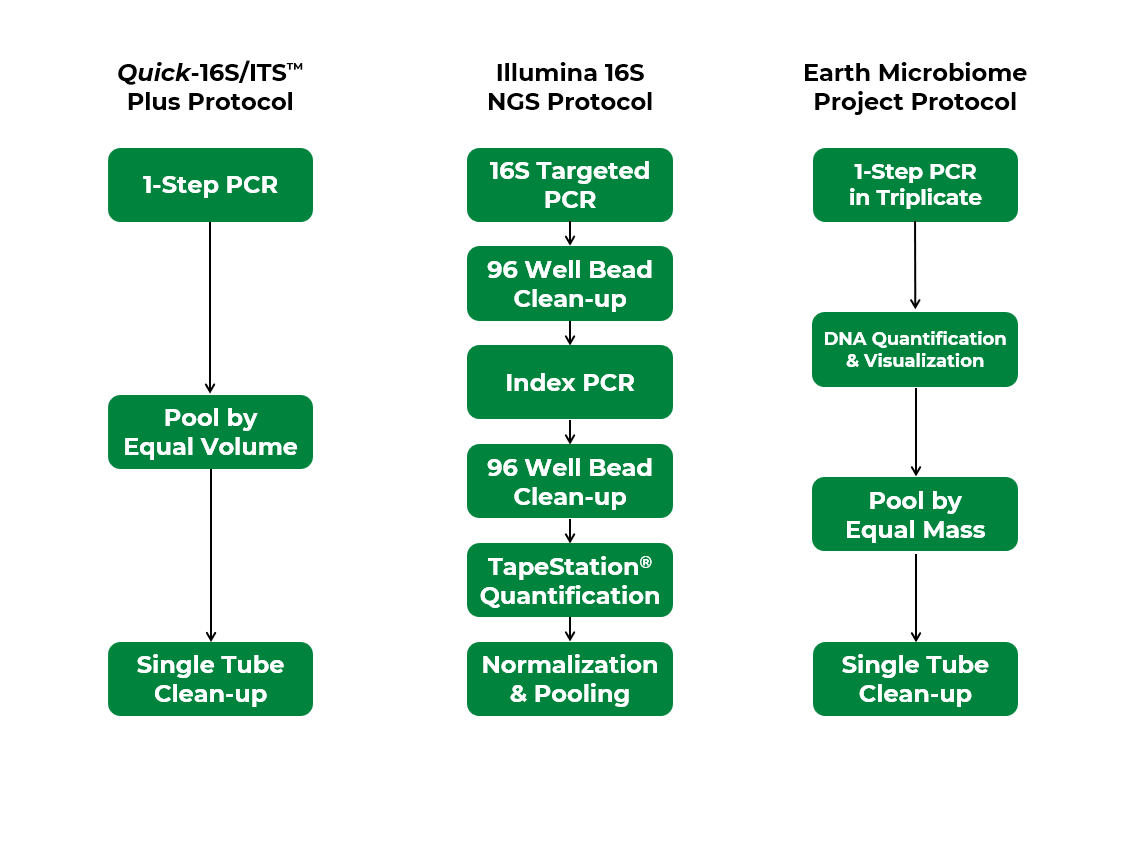
Figure 1. Workflow comparison among Quick-16S™ Plus kit, Illumina 16S protocol, and Earth Microbiome Project protocol.
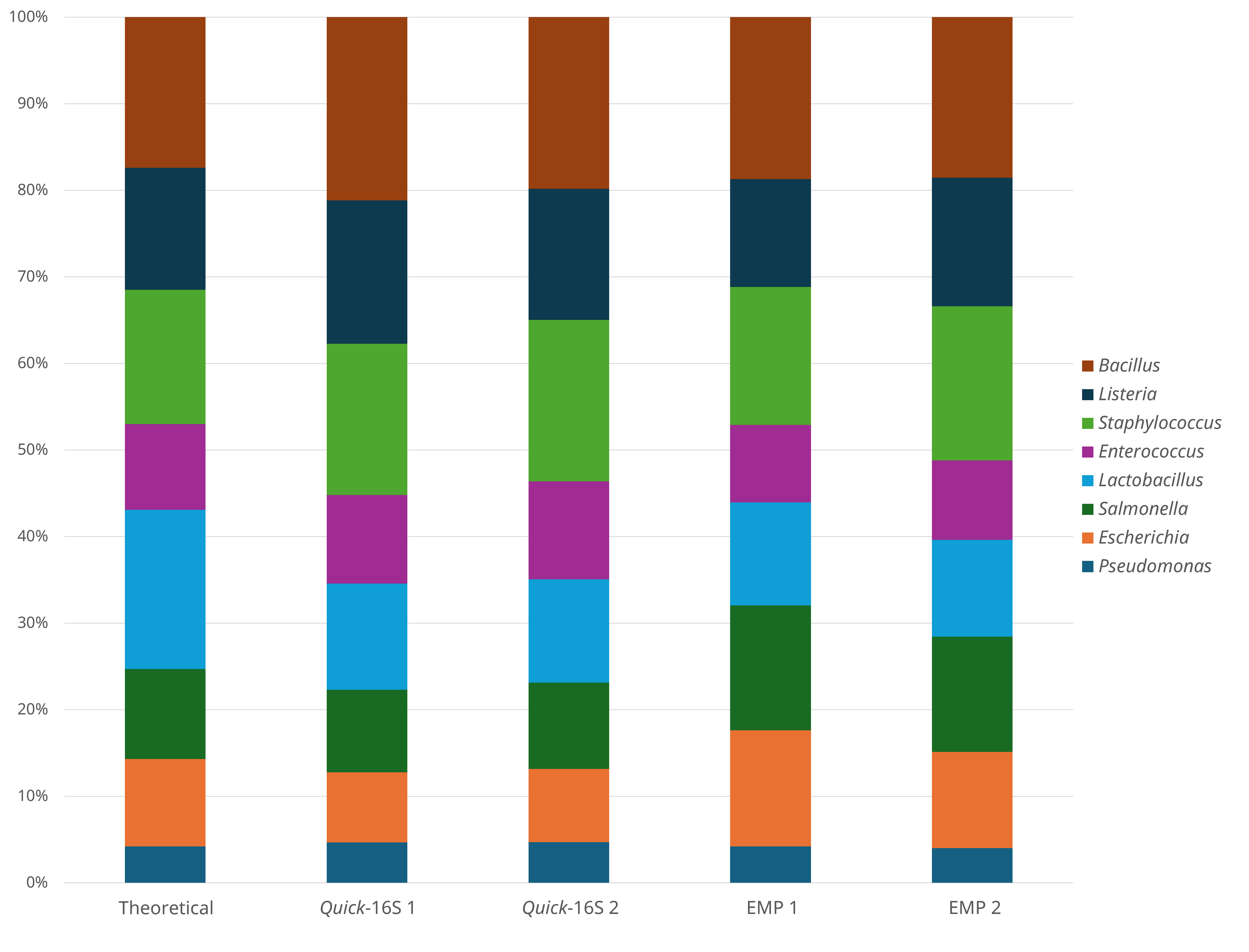
Figure 2. Comparison of microbial standards profile between Quick-16S™ Plus kit (V4) and Earth Microbiome Project protocol.
When comparing microbial profiling results, we found that both methods generated profiles closely matching the theoretical microbial composition, indicating comparable accuracy (Figure 2). A variety of additional sample types were also tested using both methods – including feces, soil, and river sediment. The Quick-16S™ Plus kit consistently showed lower variation in total numbers of reads generated per library, as measured by the coefficient of variation (CV), indicating better normalized read counts across samples compared to the EMP protocol (Table 1). Additionally, we observed that the Quick-16S™ Plus kit generated a smaller percentage of chimeric reads among various sample types (Table 2), addressing a common challenge inherent to 16S amplicon sequencing caused by PCR amplification.6 When analyzing microbial profiles across all three sample types, both methods produced comparable results (Figure 3).
| Quick-16S Plus | EMP | |
|---|---|---|
| Fecal (n=6) | 9.74% | 21.71% |
| River Sediment (n=6) | 9.96% | 26.62% |
| Soil (n=6) | 8.91% | 19.28% |
| All (n=18) | 9.92% | 26.46% |
| Quick-16S Plus | EMP | |
|---|---|---|
| Fecal (n=6) | 1.01% | 12.48% |
| River Sediment (n=6) | 1.28% | 1.92% |
| Soil (n=6) | 1.41% | 1.73% |
| All (n=18) | 1.23% | 4.99% |
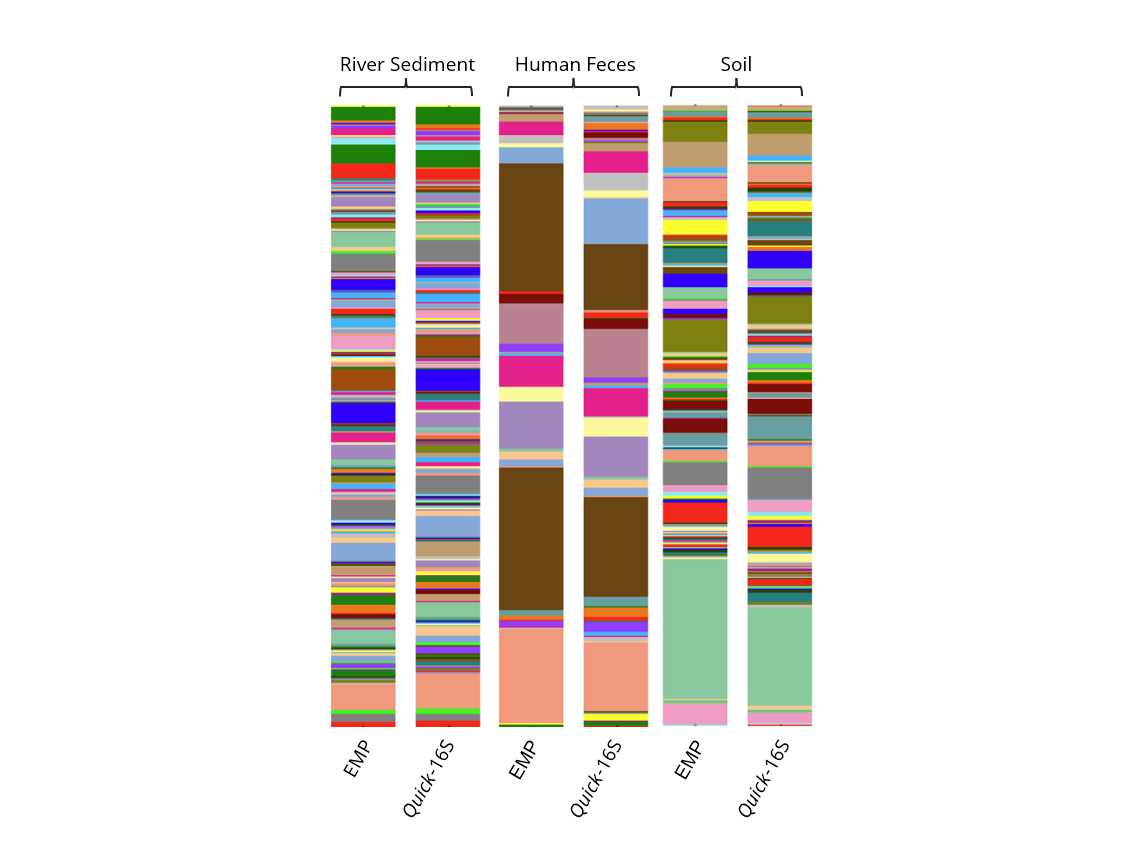
Figure 3. Comparison of biological and environmental sample profile between Quick-16S™ Plus kit (V4) and Earth Microbiome Project protocol
In summary, the Quick-16S™ Plus NGS Library Prep Kit (V4) requires fewer steps, accurately profiles microbial communities, produces more consistent read counts across diverse samples, and generates fewer chimeric reads, making it a streamlined and effective alternative to the EMP protocol.
Next, we compared the performance of the Quick-16S™ Plus NGS Library Prep Kit (V3-V4)5 to the Illumina 16S protocol (V3-V4) by sequencing the ZymoBIOMICS® Microbial Community DNA Standard and a variety of biological and environmental samples. The Illumina protocol requires six steps —twice as many as the three steps needed by the Quick-16S™ Plus kit (Figure 1). Specifically, the Illumina protocol involves two separate PCR reactions and 96-well bead clean-ups, as well as individual quantification and normalization of each sample. In contrast, the Quick-16S™ Plus kit simplifies the workflow to a single PCR step and a single-tube bead cleanup, while eliminating the need for both quantification and normalization (Figure 1). When comparing microbial profiling results, both methods yielded microbial profiles closely aligned with the theoretical profile, indicating comparable accuracy (Figure 4).
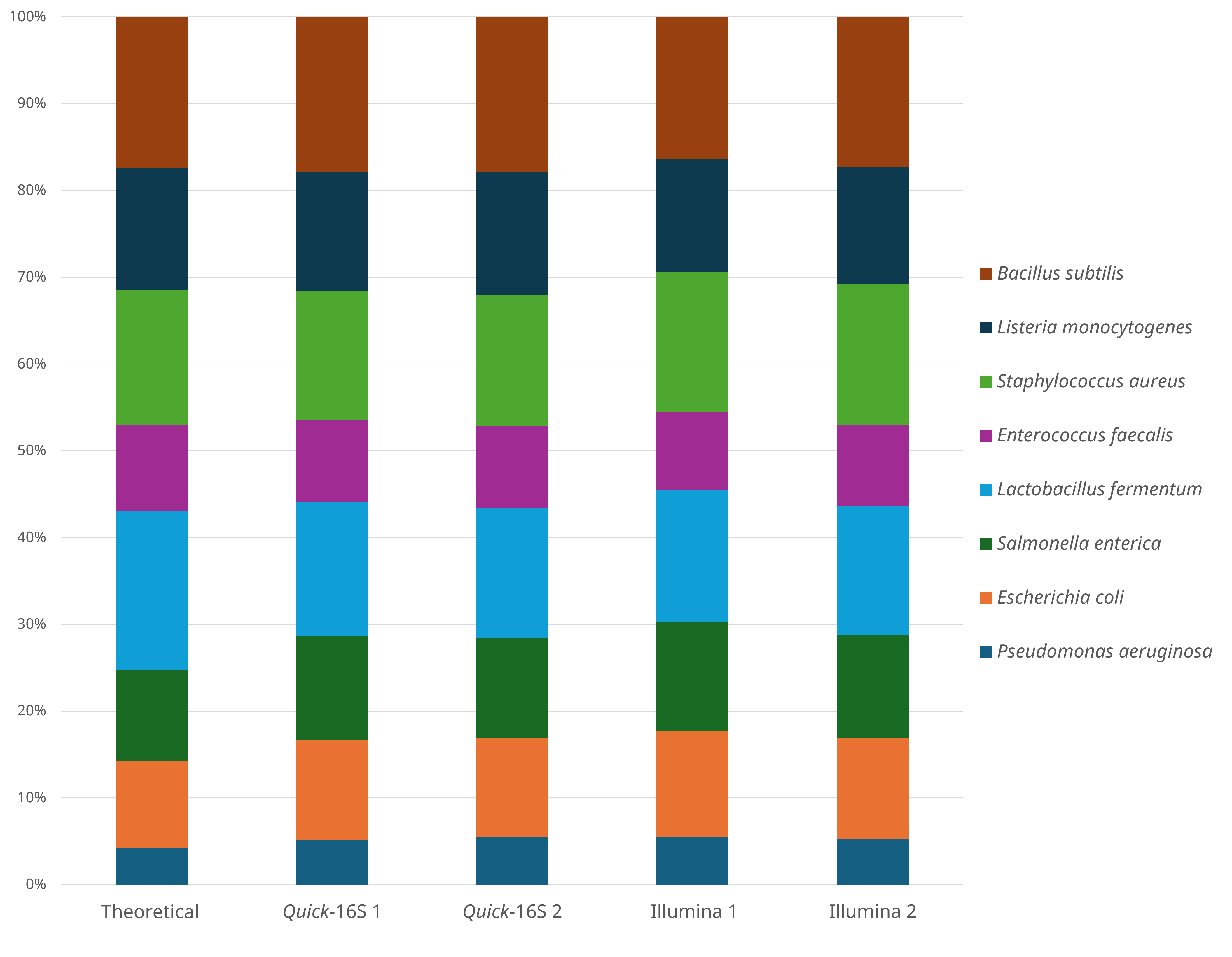
Figure 4. Comparison of microbial standards profile between Quick-16S™ Plus kit (V3-V4) and Illumina 16S protocol.
We again applied both methods to a variety of sample types, including human and horse feces, and soil samples. The Quick-16S™ Plus kit showed lower variation in total numbers of reads generated per library compared to the Illumina 16S protocol, as measured by the coefficient of variation (CV) (Table 3).
| Quick-16S Plus | Illumina | |
|---|---|---|
| Horse Fecal (n=10) | 9.63% | 6.29% |
| Human Fecal (n=10) | 13.01% | 20.20% |
| Soil (n=10) | 13.50% | 153.99% |
| All (n=30) | 24.5% | 57.23% |
Notably, the Illumina 16S protocol was observed to have underrepresented archaeal taxa in horse fecal samples (Figure 5), likely due to its primers being optimized for bacterial sequences rather than those of archaea.7 In contrast, the primers used in the Quick-16S™ Plus kit are designed to amplify both bacterial and archaeal sequences, resulting in a significantly higher proportion of archaeal reads being captured (Figure 5).
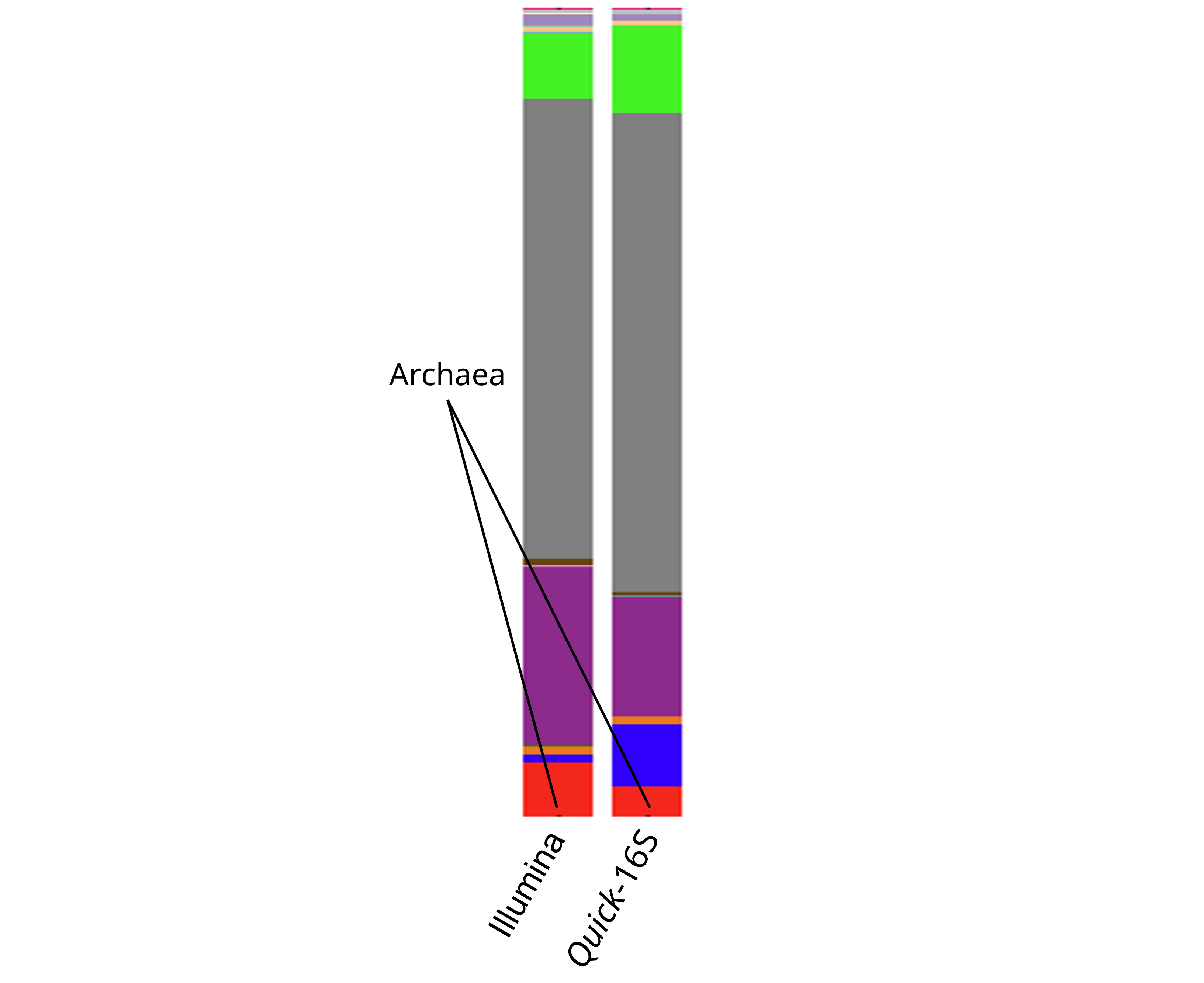
Figure 5. Comparison of horse feces composition between Quick-16S™ Plus kit (V3-V4) and Illumina 16S protocol
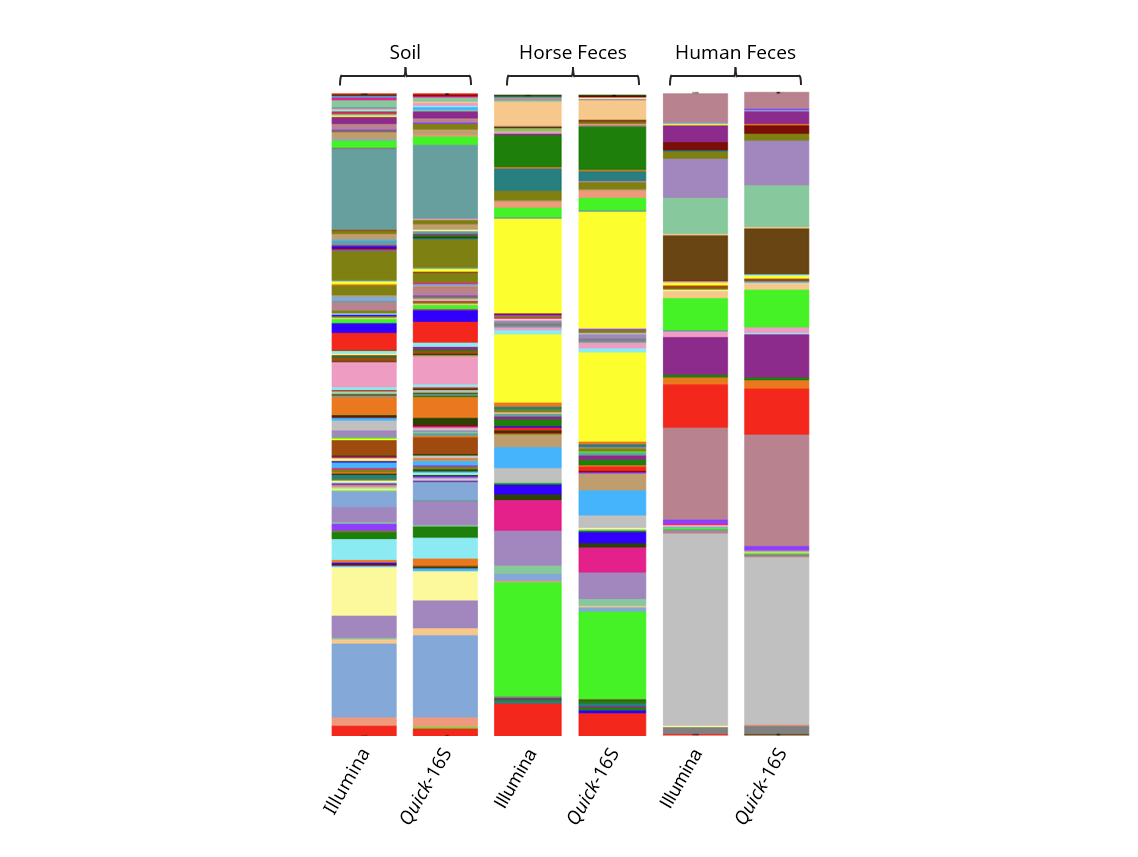
Figure 6. Comparison of profile between Quick-16S™ Plus kit (V3-V4) and Illumina 16S protocol across a variety of biological and environmental samples.
| Quick-16S Plus | Illumina | |
|---|---|---|
| Horse Fecal (n=10) | 1.40% | 2.05% |
| Human Fecal (n=10) | 3.58% | 2.09% |
| Soil (n=10) | 0.48% | 0.84% |
| All (n=30) | 1.65% | 1.89% |
Additionally, we observed that Zymo kit generated a smaller percentage of chimeric reads among various sample types (Table 4). Finally, when examining the microbial profiles, we found that both methods produced comparable results across all three sample types (Figure 6). In summary, the Quick-16S™ Plus NGS Library Prep Kit (V3-V4) requires fewer steps, maintains equal accuracy, generates consistent read counts, avoids underrepresentation of archaeal reads, and generates fewer chimeric reads, making it a streamlined and effective alternative to Illumina’s 16S NGS protocol.
Methods
DNA Extraction
Samples were collected and stored in DNA/RNA Shield™, following the storage recommendations for each sample type. DNA was extracted using the ZymoBIOMICS® 96 MagBead DNA Kit. DNA was stored frozen at -20°C until ready for library preparation.
Earth Microbiome Project Protocol
Targeted PCR amplification was performed on the V4 region of the 16S rRNA gene using the primer pair 515f and 806r with barcode sequences on the forward primer. Each 25 µL PCR reaction contained 13 µL of PCR-grade water, 10 µL of 2X Platinum Hot Start PCR Master Mix, 0.5 µL of 10 µM forward primer, 0.5 µL of 10 µM reverse primer, and 1 µL of sample. The thermocycler conditions for PCR were as follows: 94°C for 3 minutes to denature DNA, with 35 cycles of 94°C for 45 seconds, 50°C for 60 seconds, 72°C for 90 seconds; with a final extension of 72°C for 10 minutes to ensure complete amplification. Samples were prepared in triplicate and each of the replicates were combined into a single tube post-amplification. To verify the presence of PCR product, the samples were run using an Agilent Tapestation™ 4150 with High Sensitivity D1000 ScreenTape. Samples were quantified via Qubit 3.0 Fluorometer, using a Qubit dsDNA HS Assay Kit, and 240 ng of PCR product from each sample was pooled into a single tube. The amplicon pool was cleaned using the DNA Clean & Concentrator-5. Amplicons were sequenced on an Illumina MiSeq™ at 2x150bp using a MiSeq™ Reagent Nano Kit v2 (300-cycles) and sequencing primers and procedures as described in the Earth Microbiome Project protocol.
Illumina Metagenomic Protocol
Targeted PCR amplification of the V3-V4 region was performed on each sample using 341f and 806r primers with overhang adapter sequences. Each 25 µL PCR reaction contained 12.5 µL of 2X KAPA HiFi HotStart Ready Mix, 5 µL of 1 µM forward primer, 5 µL of 1 µM reverse primer, and 2.5 µL of sample at a concentration of 5 ng/µL. The thermocycler conditions were as follows: 95°C for 3 minutes to denature DNA, with 25 cycles of 95°C for 30 seconds, 55°C for 30 seconds, and 72°C for 30 seconds; with a final extension of 72°C for 5 minutes to ensure complete amplification. Following PCR amplification, amplicons were cleaned using Select-a-Size™ Magbeads at a ratio of 0.8x. The cleaned amplicons were barcoded using Illumina Nextera™ XT Index primers. The index PCR set up was as follows: 25 µL of 2X KAPA HiFi HotStart ReadyMix, 5 µL of Nextera™ XT Index Primer 1, 5 µL of Nextera™ XT Index Primer 2, 10 µL of PCR-grade water, and 5 µL of cleaned amplicons. The thermocycler conditions were the same as the amplicon PCR above with 8 cycles of amplification instead of 25 cycles. Following index PCR, barcoded libraries were cleaned using Select-a-Size™ Magbeads at a ratio of 0.8x. The libraries were visualized and quantified via Agilent Tapestation™ 4150 High Sensitivity D1000 ScreenTape and Qubit dsDNA HS Assay. Libraries were pooled based on equal molarity and sequenced on an Illumina NextSeq™ 2000 at 2x300bp using an Illumina NextSeq™ 1000/2000 P1 Reagent (600-cycles) kit.
Quick-16S™ Plus Kit Protocol
Libraries targeting the V4 region and the V3-V4 region of the 16s rRNA gene were prepared using the Quick-16S™ Plus NGS Library Prep Kit (V4) [D6430] and Quick-16S™ Plus NGS Library Prep Kit (V3-V4) [D6421]. Briefly, 2 µL of DNA sample was added to 8 µL of reaction premix and amplified via real-time thermocycler. The thermocycler conditions were as follows: 95°C for 10 minutes and 42 cycles of 95°C for 30 seconds, 55°C for 30 seconds, 72°C for 3 minutes. Equal volumes of PCR product were pooled into a single tube and the resulting pool was cleaned using Select-a-Size™ Magbeads at a ratio of 0.8x. The V3-V4 libraries were sequenced on an Illumina NextSeq™ 2000 at 2x300bp using an Illumina NextSeq™ 1000/2000 P1 Reagent (600-cycles) kit. The V4 libraries were sequenced on an Illumina MiSeq™ at 2x150bp using a MiSeq™ Reagent Nano Kit v2 (300-cycles) with addition of the custom sequencing primers provided in the kit.
Bioinformatics
Species-level identification from next generation sequencing data was performed using an in-house pipeline offered by Zymo Research Microbiome Sequencing Service that utilizes DADA28 for taxonomic classification and QIIME 29 for read filtering.

Citations
- Cao, Yu, et al. "A review on the applications of next generation sequencing technologies as applied to food-related microbiome studies." Frontiers in Microbiology8 (2017): 1829.
- Botan, Alexandru, et al. "Performance of 16S rRNA Gene Next-Generation Sequencing and the Culture Method in the Detection of Bacteria in Clinical Specimens." Diagnostics13 (2024): 1318.
- Bharti, Richa, and Dominik G. Grimm. "Current challenges and best-practice protocols for microbiome analysis." Briefings in Bioinformatics1 (2021): 178-193.
- https://earthmicrobiome.org/protocols-and-standards/16s/
- https://support.illumina.com/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf
- Gohl, Daryl M., et al. "Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies." Nature biotechnology 34.9 (2016): 942-949.
- Zhao, Jun, Jonathan Rodriguez, and Willm Martens-Habbena. "Fine-scale evaluation of two standard 16S rRNA gene amplicon primer pairs for analysis of total prokaryotes and archaeal nitrifiers in differently managed soils." Frontiers in Microbiology 14 (2023): 1140487.
- Callahan, Benjamin J., et al. "DADA2: High-resolution sample inference from Illumina amplicon data." Nature methods7 (2016): 581-583.
- Bolyen, Evan, et al. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. No. e27295v1. PeerJ Preprints, 2018.