What is RNA-Seq? Introduction, Benefits, and Workflow
The scope of Next-Generation Sequencing (NGS) and RNA sequencing (RNA-seq) are rapidly evolving and expanding.
What is RNA Sequencing (RNA-Seq)?
The growing field of RNA sequencing (RNA-Seq) seeks to elucidate the many roles of RNA in the modulation of cellular and biological processes, not only as an intermediate molecule but in its other functions. RNA-Seq provides both qualitative and quantitative information about the different RNA species present in a sample, enabling researchers to better understand the RNA environment at different biological, physiological, or pathological states.
There are many different RNA sequencing methods, each optimized for different applications. To name a few examples:
- Total RNA-Seq which profiles both coding and noncoding RNAs present in a sample. It commonly is performed with ribosomal RNA depletion.
- mRNA-Seq, including 3′ mRNA-Seq, which produces a summary of protein-coding gene expression from eukaryotic samples.
Why use RNA Sequencing (RNA-Seq)?
Prior to RNA-Seq, the main technology for RNA analysis and gene expression profiling was microarrays. The main limitation of this technology is that it can only detect predefined RNA sequences, providing a biased view of the transcriptome. RNA-Seq is not limited to known sequences and produces a significantly larger data output. In addition to gene expression analysis, RNA-Seq can determine novel transcripts, alternative splice variants, single nucleotide polymorphisms (SNPs), insertions/deletions, and other RNA variations.
A typical RNA-Seq sample prep workflow includes steps of sample collection, RNA extraction, library preparation, sequencing, and bioinformatics analysis. Depending on the specific type of RNA-Seq and the desired analyses, certain aspects of the workflow can vary significantly. To provide an overview of how to perform RNA sequencing, we will briefly discuss the significance of each step of the workflow, as well as key aspects of library preparation.
Step-by-Step RNA-Seq Sample Prep Workflow
Sample Collection and RNA Extraction
To ensure an accurate view of the transcriptome, it is essential that the RNA is preserved at the initial step of sample collection. Stabilization reagents such as DNA/RNA Shield can be utilized to store samples, protecting the integrity of the RNA until extraction. Most RNA-Seq methods benefit from using RNA that is DNA-free and of high integrity and purity, so select an extraction method that can reliably process your specific sample type while including a DNA removal step such as DNase I treatment. It also must be ensured that the extraction protocol will capture your RNAs of interest. Zymo Research’s Quick-RNA kits and Direct-zol RNA kits can produce DNA-free RNA of high quality that is suitable for RNA-Seq workflows. Please see our blog “Evaluating Quality of Input RNA for NGS Library Preparation” for more information and tips to obtain RNA for use in RNA-Seq experiments.
RNA-Seq Library Preparation
Library preparation readies extracted RNA for sequencing. For Next-Generation Sequencing (NGS) based RNA-Seq, this typically involves reverse transcription of the RNA sample to produce cDNA, ligation of sequencing adapters to the cDNA, and indexing PCR to amplify the sample and incorporate index barcodes. The final product is a cDNA library. It is important to consider the RNA of interest and the desired types of analyses when selecting a library preparation method, as the specific mechanisms of each can make it suitable for different goals. Our blog, “Considerations for Choosing between Total RNA-Seq and 3′ mRNA-Seq”, provides further information on two common library preparation methods. Additionally, it is critical to ensure the compatibility of the produced libraries with the sequencing instrument that will be utilized.
Accommodating Abundant RNA Species
An important factor is how the library preparation method deals with overly abundant molecules in an RNA sample, such as ribosomal RNA (rRNA). Total RNA can comprise up to 80-95% rRNA by mass in eukaryotes. These abundant transcripts will consume the vast majority of reads during a sequencing run if unaccounted for. If the experimental goal is not examining rRNA, these reads will be uninformative and overshadow the other RNAs of interest, wasting money and resources. There are several techniques to limit the prevalence of rRNA molecules. These include rRNA depletion and target enrichment.
rRNA Depletion
rRNA depletion describes the procedure that removes rRNA from a total RNA sample. rRNA depletion is typically used with total RNA-Seq, which examines both coding and non-coding RNAs. Two main types of strategies are implemented to achieve rRNA depletion: probe-based and enzymatic-based.
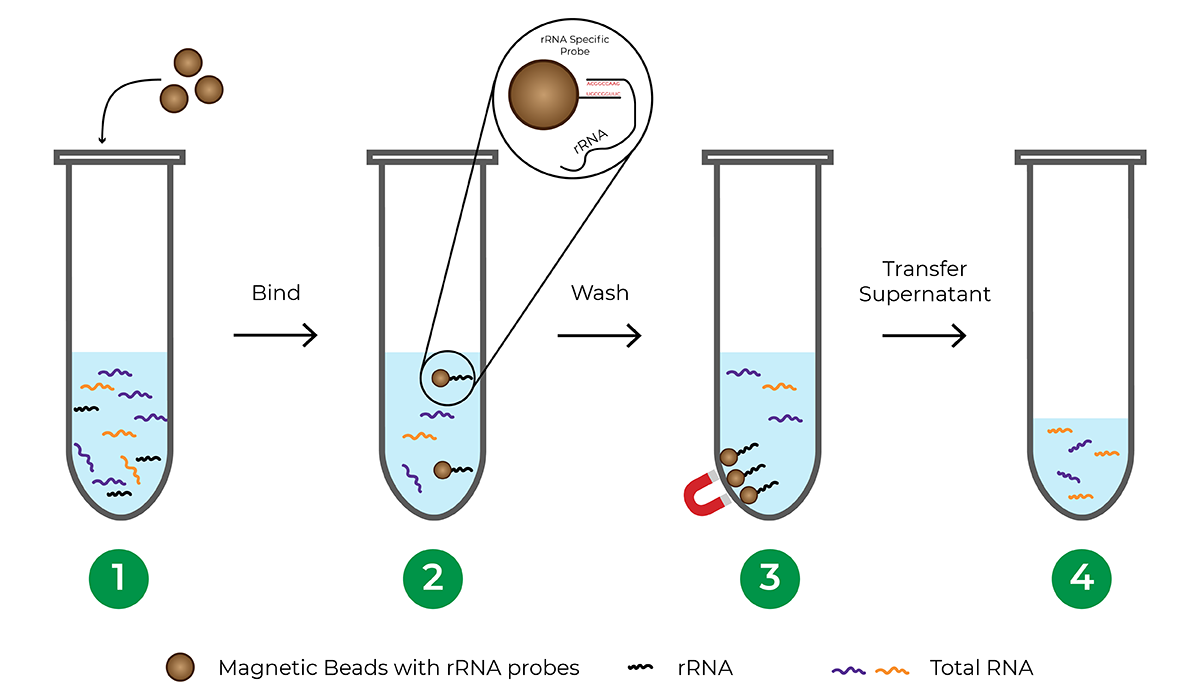
How does probe-based rRNA depletion work?
Probe-based rRNA depletion commonly utilizes beads, usually magnetic, that are functionalized with DNA probes complementary to rRNA sequences. Once the rRNA is hybridized to the probes, the beads are separated from the solution, leaving an rRNA-depleted total RNA sample.
The major limitation of probe-based depletion is that each species requires its own probe panel to effectively deplete rRNA from the sample since rRNA sequences vary between different organisms. Probe panels are commercially available for commonly studied model organisms. However, many species including less-studied and non-model organisms do not have probe panels available, requiring significant investment of time and money to develop a custom probe panel. Another factor to consider when using a bead-based hybridization procedure is that the process typically involves a lengthy incubation period, sometimes requiring overnight incubation.
How does enzymatic rRNA depletion work?
Instead of probes, enzymatic rRNA depletion takes advantage of the hybridization kinetics between the RNA and cDNA. Because of the abundance of rRNA, it hybridizes to its cDNA with higher efficiency after denaturation than other RNA types. In this hybridized state, the rRNA-cDNA duplex is susceptible to degradation from double-stranded nucleases. cDNAs from other RNAs such as mRNA and ncRNA are more likely to remain single stranded, thus maintaining their presence in the sample for subsequent library preparation.
The major benefit of this probe-free approach is that one kit can deplete rRNA from any organism! This universal compatibility is especially beneficial for projects involving less studied and non-model organisms where probes are expensive or not readily available. Research labs or core facilities that carry out library preparation for many species can also benefit from a streamlined workflow using one method to deplete rRNA from all of their samples. The Zymo-Seq RiboFree Total RNA Library Kit is an all-inclusive library preparation kit that integrates a novel probe-free, enzymatic rRNA depletion strategy, enabling universal compatibility with any organism. The RiboFree® technology has been applied to a wide range of research applications including, but not limited to, functional annotation of animal genomes1, lncRNA landscape profiling for neuroscience2, host plant responses to viral infection3, microbial transcriptome profiling and metatranscriptomics4-6, and pathogen detection7-10.

Target Enrichment
Target enrichment is another strategy used in certain library preparations. Unlike rRNA depletion which is subtractive, only the RNA of interest is enriched and amplified in the strategy of target enrichment, leaving other RNA at near background levels. An example of target enrichment is in the context of mRNA-Seq, including 3′ mRNA-Seq. Eukaryotic mRNA contains poly-A tails which can be targeted using oligo-dT primers. In most 3′ mRNA-Seq protocols, oligo-dT primers are used to reverse transcribe cDNA only from the mRNA, omitting the rRNA. This method does not enrich other types of RNA that lack poly-A tails, and most sequencing reads will be biased to the 3′ end of the mRNA. However, depending on the experimental goals these traits may be beneficial. The Zymo-Seq SwitchFree 3′ mRNA Library Kit provides an example library preparation method that utilizes target enrichment to allow efficient expression analysis of coding genes.
Adapter Ligation and Indexing
As part of library preparation, after obtaining cDNA with overly abundant RNA accommodated, sequencing adapters are added to the cDNA using direct ligation or other strategies. For the context of this blog, we will assume the library preparation and sequencing will be for an Illumina instrument. Adapters are short, synthetic oligonucleotides that serve two main functions. The first is to provide binding sites for flow cell hybridization and the second is to provide priming sites for sequencing.
After adapterization, the library is typically amplified and indexed via indexing PCR. The index barcode labels each library with a unique sequence so that the reads can be distinguished when multiple libraries are pooled and sequenced simultaneously via multiplexing. Different types of indexes are available depending on the specific kit, for example unique dual indexes (UDIs), the Illumina recommended indexing solution. Library preparation kits may also incorporate other types of barcodes, such as unique molecular identifiers (UMIs). While UDIs and UMIs have similar acronyms, they carry out different functions and provide distinct benefits to RNA-Seq protocols. For more information, please see our blog, “What Are UDIs and UMIs and Their Benefits in Next-Generation Sequencing?”
Quality Control, Sequencing, and Analysis
After adapter ligation and indexing the libraries are almost ready for sequencing. Quality control should be performed to ensure they are of the proper size and of sufficient concentration. This can include the use of automated electrophoresis instruments such as TapeStation or Fragment Analyzer for size characterization, as well as Qubit or qPCR-based assays for quantification. If adapter dimers are prominent, it is highly recommended to perform a size-selection cleanup to remove them. Libraries are pooled together, typically in an equimolar fashion for even read distribution. A final quantification of the pool’s concentration via a qPCR-based method is highly recommended for accurate loading onto the sequencer. Your sequencing provider usually has further details on recommendations and sample requirements.
After sequencing, the raw data is demultiplexed based on the index information and typically output in a FASTQ format for each library. This is a plain text file where the sequences are reported as single character representations of the four nucleotides, with each base call given a quality score that reflects the accuracy. The subsequent analysis commonly includes a trimming step to remove poor quality bases and superfluous adapter sequences, and an alignment step to align the trimmed reads to a reference genome. Depending on the type of RNA-Seq method used and the goals of the project, differential gene expression (DGE) analysis, alternative splicing analysis, isoform identification, and more types of analyses can be employed. Many bioinformatic tools for these analyses are open source, providing great resources for researchers to derive insights from their RNA-Seq data.
RNA-Seq is a powerful tool to expand knowledge of the transcriptome and its impact on life processes. NGS-based protocols are widely available, each tuned to answer different questions. Zymo Research provides complete workflows for your RNA-Seq needs, with products and services available for sample collection, RNA extraction, RNA-Seq library preparation, sequencing, and bioinformatics analysis. Learn more about our innovative technologies for transcriptomics, including RNA-Seq library kits and RNA-Seq services (with free consultation included).
References
- Overbey, E. G.; Ng, T. T.; Catini, P.; Griggs, L. M.; Stewart, P.; Tkalcic, S.; Hawkins, R. D.; Drechsler, Y. Transcriptomes of an Array of Chicken Ovary, Intestinal, and Immune Cells and Tissues. Front Genet 2021, 12, 664424. DOI: 10.3389/fgene.2021.664424.
- Talross, G. J. S.; Carlson, J. R. The rich non-coding RNA landscape of the Drosophila antenna. Cell Rep 2023, 42 (5), 112482. DOI: 10.1016/j.celrep.2023.112482.
- Kanodia, P.; Miller, W. A. Effects of the noncoding subgenomic RNA of red clover necrotic mosaic virus in virus infection. J Virol 2021, JVI0181521. DOI: 10.1128/JVI.01815-21.
- Onetto, C. A.; Costello, P. J.; Kolouchova, R.; Jordans, C.; McCarthy, J.; Schmidt, S. A. Analysis of Transcriptomic Response to SO2 by Oenococcus oeni Growing in Continuous Culture. Microbiol Spectr 2021, 9 (2), e0115421. DOI: 10.1128/Spectrum.01154-21.
- Willemse, D.; Moodley, C.; Mehra, S.; Kaushal, D. Transcriptional Response of Mycobacterium tuberculosis to Cigarette Smoke Condensate. Front Microbiol 2021, 12, 744800. DOI: 10.3389/fmicb.2021.744800.
- Malone, M.; Radzieta, M.; Peters, T. J.; Dickson, H. G.; Schwarzer, S.; Jensen, S. O.; Lavery, L. A. Host-microbe metatranscriptome reveals differences between acute and chronic infections in diabetes-related foot ulcers. APMIS 2022, 130 (12), 751-762. DOI: 10.1111/apm.13200.
- Wang, J.; Pan, Y. F.; Yang, L. F.; Yang, W. H.; Lv, K.; Luo, C. M.; Kuang, G. P.; Wu, W. C.; Gou, Q. Y.; Xin, G. Y.; et al. Individual bat virome analysis reveals co-infection and spillover among bats and virus zoonotic potential. Nat Commun 2023, 14 (1), 4079. DOI: 10.1038/s41467-023-39835-1.
- Wampande, E. M.; Waiswa, P.; Allen, D. J.; Hewson, R.; Frost, S. D. W.; Stubbs, S. C. B. Phylogenetic Characterization of Crimean-Congo Hemorrhagic Fever Virus Detected in African Blue Ticks Feeding on Cattle in a Ugandan Abattoir. Microorganisms 2021, 9 (2). DOI: 10.3390/microorganisms9020438.
- Keremane, M.; Singh, K.; Ramadugu, C.; Krueger, R. R.; Skaggs, T. H. Next Generation Sequencing, and Development of a Pipeline as a Tool for the Detection and Discovery of Citrus Pathogens to Facilitate Safer Germplasm Exchange. Plants (Basel) 2024, 13 (3). DOI: 10.3390/plants13030411.
- Licastro, D.; Rajasekharan, S.; Dal Monego, S.; Segat, L.; D'Agaro, P.; Marcello, A. Isolation and Full-Length Genome Characterization of SARS-CoV-2 from COVID-19 Cases in Northern Italy. J Virol 2020, 94 (11). DOI: 10.1128/JVI.00543-20.