Contents
A Guide on Bisulfite Conversion for Beginners
Overview of Bisulfite Conversion
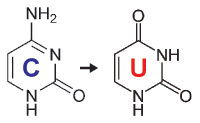
When starting with bisulfite conversion, there are a number of considerations to keep in mind to ensure optimal performance and subsequent accuracy of the DNA methylation analysis. Treatment with bisulfite is inherently a harsh process that dramatically changes both the chemical makeup and physical properties of the DNA. Input DNA transforms from a large, stable, double-stranded molecule to a collection of randomly fragmented, single-stranded fragments having almost all cytosines completely changed to uracil. These changes dramatically influence quantification, quality assessment, amplification, and analysis of the bisulfite-converted DNA. These points and the special considerations they require with respect to UV spectrophotometry (NanoDrop), agarose gel electrophoresis, and PCR are highlighted below.
Quantification of Bisulfite-Converted DNA

Converted DNA should be quantitated as RNA using a UV spectrophotometer (NanoDrop) with Ab260 nm 1.0 = 40 µg/ml. When determining the recovery of the bisulfite-converted DNA, two primary factors need to be considered 1.) The intactness of the starting material, and 2.) RNA contamination. The quality of the DNA, in terms of size, used as input for bisulfite conversion is the most important factor when assessing recovery. Degraded starting material will lead to increased sample loss during the bisulfite conversion process. Additionally, RNA contamination will contribute significantly to the Ab260 nm resulting in an overestimation of DNA quantity. RNA carried over into the conversion is lost making the yields appear low compared to the input. Importantly, the conversion is not compromised in either of the above cases.
Assessing Quality of Bisulfite-Converted DNA
Agarose gel electrophoresis (2% gel with 100 bp marker) of converted DNA can be used to gauge recovery and fragmentation. Most often nothing will be visible in the gel when it is first removed from the gel tank, this is normal and results from the DNA being almost entirely single stranded following the conversion. Chilling the gel for several minutes in an ice bath will force enough base-pairing to allow intercalation of the ethidium bromide for the DNA to be visible. The converted DNA will run as a smear, generally from > 1,500 down to 100 bp. It is important that enough material be loaded into the gel to allow visualization: typically about 100 ng/well is sufficient.


The Challenges of Bisulfite-Converted DNA - DNA fragmentation and limited base pairing makes it difficult to see the DNA following bisulfite conversion. Samples of bisulfite converted human genomic DNA were run in a 2% agarose gel with a 100 bp marker visualized normally (left panel) and after chilling in an ice bath (right panel).
Bisulfite PCR and Primer Design
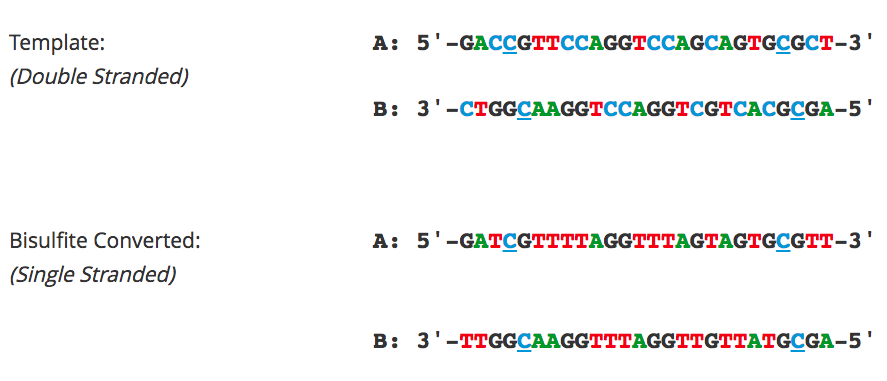
Bisulfite PCR is the most common technique used for methylation analysis of bisulfite-converted DNA and also the most susceptible to mishap. Keep in mind the DNA will be significantly fragmented, as the strands are no longer complementary, and almost completely devoid of cytosine. Primer design is the key to successful bisulfite PCR. Unlike normal PCR, bisulfite PCR primers need to be long (usually between 26-30 bases) and the amplicon size should be relatively short (between 150-300 bp). Ideally, the primers should not contain CpG sites, however, if they are necessary locate them at the 5'-end of the primer with a mixed base at the cytosine position. It is also important to note that only one strand of the bisulfite-converted template will be amplified by any given primer set. Only the reverse primer will actually bind to the target DNA, which in turn will generate template for the forward primer to anneal. Usually, 35 to 40 cycles are required for successful amplification. Hot start polymerases are strongly recommended as non-specific amplification is relatively common with bisulfite-converted DNA due to it being AT-rich. Annealing temperatures between 55-60°C typically work well, and an annealing temperature gradient should be run with every new primer set to ensure optimal amplification of the specific target.
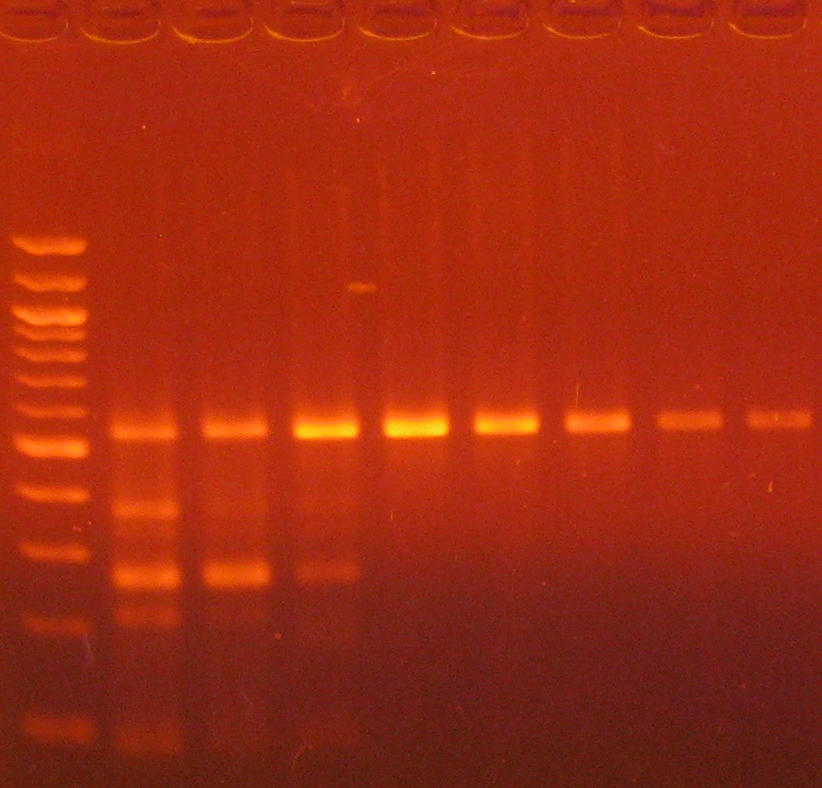
Annealing Gradient for Primers used for Bisulfite PCR.Replicate bisulfite PCRs were incubated in a gradient thermal cycler using an annealing temperature gradient of 55 to 65oC (left to right). Equal volumes from each reaction were analyzed in a 2% agarose gel with a 100 bp DNA marker.
Primer Design for Methylation Specific PCR (MSP)
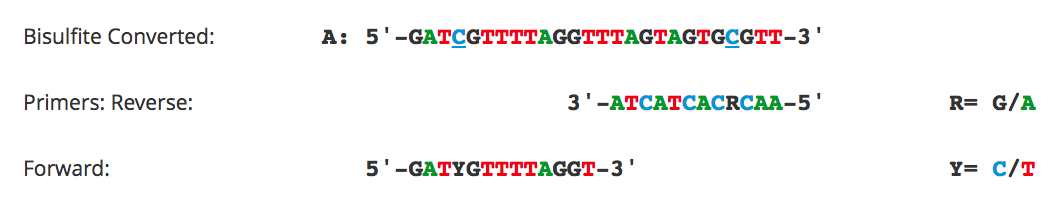
Methylation Specific PCR (MSP) relies on amplification to assess the methylation status at specific CpG sites. Success with this system depends on the differential amplification of the template using methylated (M) and non-methylated (U) primer sets. While most of the considerations for primer design are identical to those for bisulfite PCR, treatment of CpG sites within the primer is completely different. For MSP it is necessary to locate CpG sites at the 3'-end of the primers with cytosines in the methylated (M) primers and thymines in the non-methylated (U) primers.
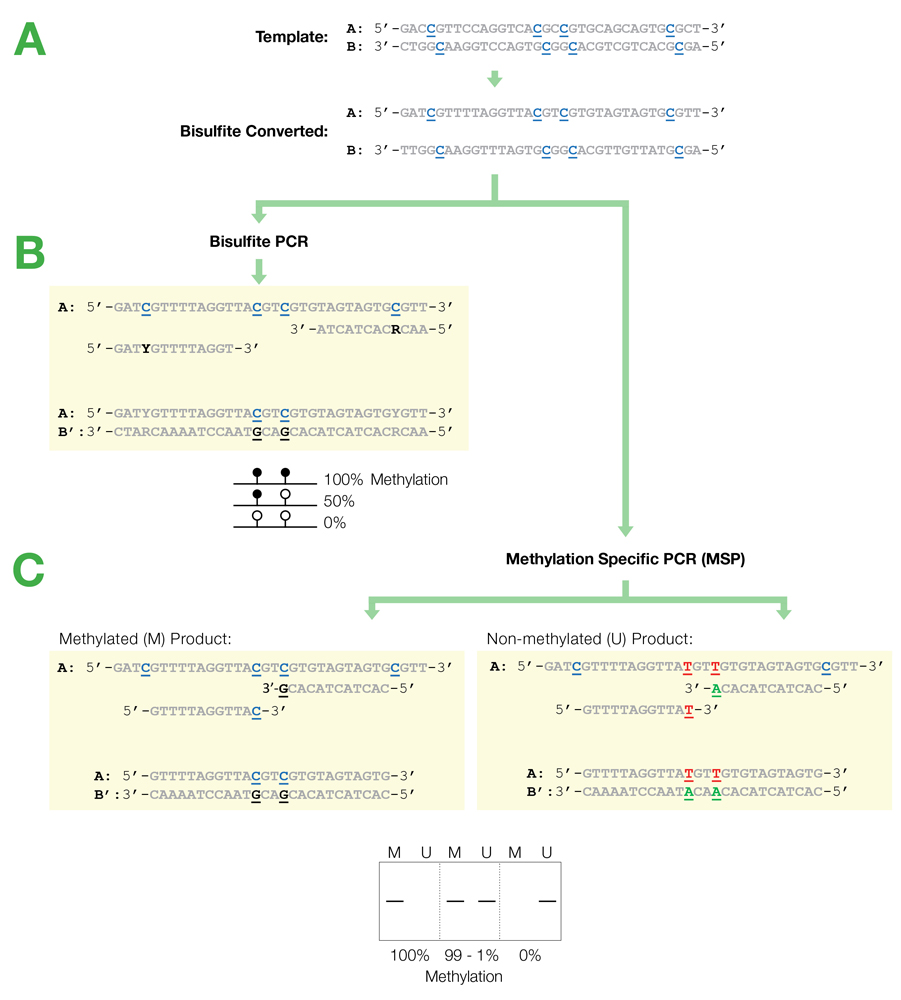
Flowchart of primer design for bisulfite PCR and Methylation Specific PCR (MSP). (A) Following bisulfite treatment, the two converted strands of the DNA template are no longer complementary. It is important to note that only one strand will be amplified with any given primer set. This is illustrated by the following examples: (B) Primers for Bisulfite PCR are designed for subsequent sequencing and analysis of cytosines within the amplicon. CpG sites within the primers should be avoided or located at the 5'-end with a mixed base at the cytosine position (Y= C/T, R= G/A). Sequencing data is commonly represented by a “lollipop” plot where closed circles represent methylated cytosine positions and open circles non-methylated ones. (C) Primers for Methylation Specific PCR (MSP) are designed to target and assess the methylation status at specific CpG sites. CpG sites within the primers must be located at the 3’-end to increase their specificity to methylated (M) or non-methylated (U) templates. Completely methylated or non-methylated templates will generate a single amplicon from only their representative primer set following MSP. Samples with mixed methythation, will be amplified by both primer sets.
Bisulfite Sequencing
Bisulfite sequencing is still among the most common techniques used for analyzing bisulfite-converted DNA and provides single base resolution across the entire amplicon. Cloning followed by sequencing with vector-specific primers is recommended to obtain the best sequencing results for quantification of methylation. Direct sequencing of bisulfite PCR products is not recommended as it generally yields poor sequencing quality and will confound quantification of partially methylated sites. Pyrosequencing can be used as an alternative to direct sequencing.
Next-Gen Bisulfite Sequencing
Next-Gen sequencing-based applications such as Reduced Representation Bisulfite Sequencing (RRBS) and Whole-Genome Bisulfite Sequencing (WGBS) are increasingly utilized to obtain genome-wide single base resolution methylation analysis and to identify putative biomarker candidates. In both workflows, conversion efficiency is of paramount concern, as even relatively small variations in efficiency can effect thousands of individual sites. If library preparation is to be performed prior to bisulfite conversion, it is important that the adaptors added to the fragmented DNA be methylated to preserve their sequence. Use of proper control DNAs are also recommended if non-CpG methylation is a factor in the samples (e.g., in plants) and to account for the inability of bisulfite conversion to differentiate between methylation (5-mC) and hydroxymethylation (5-hmC).
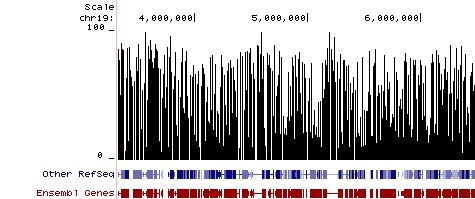
Methylation Plot from Reduced Representation Bisulfite Sequencing (RRBS). Methylation Plot from Reduced Representation Bisulfite Sequencing (RRBS). Data shows the relative percentage of methylation at individual CpG sites in mouse DNA. Methylation percentage is shown across a ~3 Mb region of mouse chromosome 19. Bisulfite sequencing libraries were prepared using mouse genomic DNA prepped with the Genomic Clean & Concentrator (D4010, D4011 - Zymo Research) and bisulfite converted using EZ DNA Methylation technology prior to Next-Gen sequencing.